QMS・GVP体制の構築と運用支援
製造販売業者は「QMS」「GVP」という基準に基づき、組織と文書を整備し、実務にあたる従業員の教育訓練・スキル管理等をしながら、管理業務を行うことになります。 製造販売業者から製造委託を受けている医療機器製造業者においても、適切な製造管理・品質管理体制を整備する必要があります。 新規に医療機器の製造販売(輸入を含む)を開始しようという事業者様は、QMS省令に対応しなければなりません。医療機器の承認・認証の維持の要件にもなっています。 また、医療機器の製造の受託をする製造業者も、QMS省令が適用になります。医療機器製造業登録を受けるのみではなく、QMS省令への対応を含めて、受託の計画と準備を進める必要があります。
QMS省令とは
QMSとは
QMSは Quality Management System 品質管理監督システムの略です。 日本の医療機器の分野においては、薬機法に基づいて定められている「QMS省令」(厚生労働省令第169号)を指すことが多いです。 QMS省令は、国際的な品質マネジメントシステム規格であるISO9001の医療機器向け規格である「ISO13485」をもとに日本独自の上乗せ規定を追加した、品質管理の基準となる省令です。 医療機器の製造販売業者から製造委託を受ける医療機器製造業者も、適切な品質管理監督システムを構築しこれに従って業務を行わなければなりません。 2014年以前の薬事法では、「GQP」という基準が製造販売業者の許可要件になっていましたが、現在はGQPは医療機器製造販売業者には適用されていません。しかし、GQPで求められていた事項は、概ねQMS省令第3章に移行し、さらに現在のQMS省令では第2章の中に一部溶け込む形になっています。
QMS体制省令とQMS省令
QMS省令はQMS(品質管理監督システム)の組織や文書、運用について定めた基準です。 クラス2以上の医療機器はQMS省令に適合していることの調査を、新規申請の際、及び承認・認証後定期的に受け、適合しなければなりません。 他方、QMS省令を運用してゆくための体制について定めた省令がQMS体制省令であり、総括製造販売責任者の配置等について定めています。こちらは、すべての医療機器製造販売業許可(第1種、第2種、第3種)の許可の要件になっています。 製造販売業許可の許可の審査を行う都道府県は、QMS体制省令に基づき、組織、管理監督者・管理責任者・総括製造販売責任者等の配置、品質管理監督システム基準書(品質マニュアル)・製品標準書等の文書の整備状況、国内品質業務運営責任者の配置とその業務の手順書内容などを確認します。 都道府県の調査により業許可を得たとしても、それは文書化状況や組織などの体制を調査したのみであり、QMSが適切に構築・運用されていることを意味するものではありません。 医療機器の認証・承認を得るためには(あるいは、定期調査等で適合を維持するためには)、QMS省令に適合しなければなりません。 QMS省令に適合しているかどうかの調査を「QMS適合性調査」といいます。 QMS適合性調査は、認証品目ならば登録認証機関、承認品目ならば医薬品医療機器総合機構(PMDA)が行います。 体制省令の調査と異なり、運用が必要な事項についてはその実施まで調査対象です。
医療機器又は体外診断用医薬品の製造管理又は品質管理の基準に関する省令
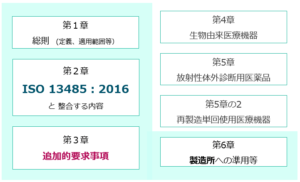
従前の省令も経過措置として2024年3月25日までは有効です。 従前の省令に基づきQMS体制を整え運用している企業は、経過措置期間中に改正QMS省令に対応しなければなりません。 改正事項の把握、要対応事項の明確化を行い、対応しましょう。 (弊所では改正QMS省令への対応について、改正事項に関するレクチャ―を含む助言・支援や文案の起案支援、運用支援、QMS適合性調査やサーベイランスのサポートを行っています)、 また、14971の改正(JIS T14971:2020)への対応(2023年9月30日まで)、ユーザビリティ規格JIS T62366-1:2019への対応等も必要となりますので、ご留意ください。 ◆QMS省令の改正への対応の助言・支援等については、こちらからお問い合わせください。
GVP省令とは
GVPとは
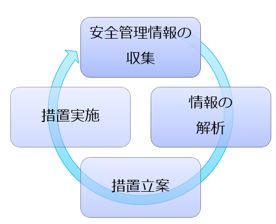
GVPは Good Vigilance Practice の略で、製造販売後の安全管理の基準です。 ユーザーや学会等から製品の安全性等に関する情報を積極的に収集し、これを解析し、対策を立て実行することが求められています。 事故の未然防止と、問題発生時の迅速な対応のための仕組みといえるでしょう。 GVP省令への適合は、製造販売業許可の要件であり、都道府県が製造販売業許可の新規の申請を受けたときや更新前に、事業者に対して調査を実施します。 また、QMS省令の中でも、情報の受付~対応等、GVPに関わる規定があるため、QMS適合性調査の中でもGVPの手順書や運用状況について調査されることがあります。
 ご相談
ご相談
QMSの適用と限定一般・限定第三種
| クラス分類 | 承認・認証・ 届出 | 審査機関 | QMS 適合 | |
|---|---|---|---|---|
| クラスIV(高度管理医療機器) | 承認 | PMDA | 必須 | |
| クラスIII | 認証基準が存在しない | 承認 | PMDA | 必須 |
| 認証基準に適合しない | PMDA | 必須 | ||
| 認証基準に適合する (=指定高度管理医療機器) | 認証 | 認証機関 | 必須 | |
| クラスII (管理) | 認証基準が存在しない | 承認 | PMDA | 必須 |
| 認証基準に適合しない | 必須 | |||
| 認証基準に適合する (=指定管理医療機器) | 認証 | 認証機関 | 必須 | |
| クラスI(一般医療機器) | 届出 | (審査なし) 提出先:PMDA | ※ | |
※クラス1の医療機器については、QMS適合性調査はありません。
限定一般医療機器・限定第三種
クラス1の医療機器のうち、「限定一般医療機器」と国が指定した医療機器(一般的名称ごとに指定)は、QMS省令のうち、適用にならない条項が多くあります。 また、限定一般医療機器のみを製造販売する製造販売業者のことを、限定第三種医療機器製造販売業者といい、QMS省令の多くの条項が適用になりません。 QMS省令に基づいて品質マニュアルや関連手順書を作成するうえで、限定一般医療機器を扱う場合や、限定第三種に該当する場合には、それに応じた文書作成が必要になります。 限定一般医療機器ではないクラス1の医療機器は、こちらをご覧ください クラス1の医療機器は、30-36条(設計開発)は適用になりません。
ご相談QMS・GVPの体制整備
これから医療機器の業許可を取得し、製品の届出をしたり認証・承認を取得したりしようという事業者の方や、 既に製造販売業許可は取得しているがQMSには対応できていない(必要な文書が整っていない、運用もできていない)という事業者様においては、これからどのようにQMSに対応すればよいのでしょうか。
要求事項の理解
まずは、QMS体制省令・QMS省令やGVP省令で、どのような仕事が求められているのかを知りましょう。 (各種書籍でもよいですし、行政書士等のサポートをうけることも有用でしょう) そして、 自社の仕事はどのようなプロセスのつながりでできているのか その仕事をどのような組織(責任者、責任と権限)で行うのか どのような手順と基準で行うのか かを検討し明確にします。 (弊所でも、サポートのご依頼をいただいた場合、これらのステップを実行します)
組織と文書
QMS省令やGVP省令では、管理監督者、(QMSの)管理責任者、総括製造販売責任者、国内品質業務運営責任者、安全管理責任者(GVP)などが登場します。 それぞれ要件があり、省令に基づく役割を担っています。 許可を取得するためにただ要件を満たす人をそろえればよいのではなく、実際の会社の指揮命令系統、部門間の連携等を勘案して、どの部署を品質部門・安全部門にするかといったことを検討してください。 仕事をするうえで組織とともに重要ななのが「文書」です。 仕事の仕方、判断基準を定めた規定・手順書や、台帳、フローチャート、帳票(フォーマット)等がこれにあたります。
品質管理監督システム基準書(品質マニュアル)、下位文書、製品標準書等の作成
医療機器の製造管理・品質管理、製造販売後安全管理を行うためには、その手順や判断基準を明確化した各種の文書を定め、それに基づいて業務を行うことが必要です。 QMSの業務の基本文書を品質管理監督システム基準書(品質マニュアル)といい、この下に、個別の業務の手順などを定めた下位文書を定めます。 個別の製品については「製品標準書」という標準書を作成しこの実現を管理してゆくことになります。 第3種医療機器製造販売業者の場合、GVPの手順書(製造販売後安全管理業務手順書)の作成は義務付けられていませんが、業務を適切に行える体制を整えるうえで 手順書またはこれに類する文書を整えることは事実上必要と考えられます。 ご相談
文書のひな型の活用……???
こうした文書類は、一部はひな形のようなものがネット上にアップロードされています。 では、医療機器の製造販売業許可取得の準備段階で、これらをダウンロードしておいておけばよいのでしょうか。 答えは「否」です。 確かにQMS省令の要求事項とISO9001は同様の規定が多いため、ISO9001のひな型も利用できるものがあるかもしれません。また、都道府県担当者による業許可の調査の段階では、実情としては、このままで通る場合もありえます。 しかし、文書の作成は、それ自体が目的なのではなく、それに基づいて業務を実施し品質管理・安全管理を行い、品質向上等につなげてゆくことが目的です。ひな形は個々の事情に合ったものではないため、ひな形のままだと現実に運用がなされないということにもつながりかねません。 (弊所ではそのような事例を数多く見てきました。 大きな回収や、薬機法違反での行政指導等につながります)
自社にあった手順書作成、そして運用を……
ですから、ただひな形をダウンロードしておいておくことが目的ではないこと、会社に見合った仕事の仕方を形作ることが重要だという点に留意していただきたいと思います。 (弊所でサポートさせていただく際には、打合せ、ヒアリングを経て、弊所のオリジナルの文書として作成または貴社での作成をサポートさせていただいております。ひな形そのままということはありません) 許可を取得することは、目的(ゴール)ではなく、それは事業者としてのスタートなのです。 自社の組織、委託先の有無、取扱製品の設計開発から出荷までの工程、製品の性質などを勘案して作成しましょう。 そして、手順書は、一度作ったら終わりでなく、内部監査・自己点検などの機会を活用して改善してゆきましょう。 認証品目であれば、認証取得以降毎年「サーベイランス」という審査又は5年目の定期調査が行われます。 承認品目は5年ごとの定期調査を受けなければなりません。 クラス1の製品についてはこうした調査はありませんが、医療機器製造販売業許可業者は、有効期間の前に、都道府県により更新調査(QMS体制の調査)が行われます。 これらの調査においては、ただ手順書が形として揃っていることだけでなく、その運用状況、いわば運用の「質」も見られます。 許可取得後こそが重要です。 (弊所では「許可後を見据えて」許可前段階から教育訓練やヒアリング、文書作成等を実施しております。) ご相談
QMSの対応~計画的に~
計画的に対応しましょう
よくある捉え方は「許認可申請は、書類を整えて役所に出せばよい」という「書類ありき」「(一過性の)手続き」という考え方です。 ただ、現実に、QMSやGVPの対応、製造販売業許可や製造業登録、製品の手続きなどをさまざまな要素が関係しますので、このような捉え方は正しいとは言えないでしょう。 医療機器を製造販売したり、承認や認証を維持したりするためには、手続きも勿論大切ですが(そしてそのために様々な準備が発生しますが)、 何より重要なのは、そうした手続き(法規制)を含めて対応できる、薬機法対応力を会社がもち、法違反状態などにならないように管理してゆくことではないでしょうか。 新規に業許可や製品の承認・認証を取得する場合でも、あるいは製造販売業許可更新等のためにQMSの求める文書を整備する場合でも、 まずは現状を把握し、要対応事項を明確化し、それに応じた計画を立てることです。
| See Share | ・法令要求事項等の理解 ・現状認識 ・社内関係者間での共有(役割認識) |
| Think | ・要対応事項の明確化 ・(基準との差分等) ・QMS省令やGVP省令の内容理解が必須) |
| Plan | ・実施計画 |
| Do | ・実施 |
早期・的確な対応の肝は、最初に現状把握・要対応事項明確化・実施計画の立案にあるかもしれません。 ※弊所では、お客様からのご相談を踏まえ、コンサルティングのような形でこれらをご提案させていただいております。 ※弊所のサポート内容は、お客様により、また製品の状況によっても、異なります。 全体の範囲と金額を一律にお客様に適用できるわけではございません。 お見積りのためにも、まずはご相談をしていただき、状況整理などをしていただくことをお勧めいたします。 ご相談
ISO9001, ISO13485との関係性
QMS省令第2章はISO13485:2003と同様の内容です。 また、そのベースになっているのは、ISO9001の考え方や規定です。 既に13485や9001に対応している事業者様がQMS省令に対応するのであれば、QMS省令との差分を明確に把握していただく必要があるでしょう。9001とは文書化要求内容が異なります。また、QMS省令は、第3章として、旧法で適用になっていたGQPの要求が盛り込まれていますので、この要求への対応も必要です。 留意事項として、QMS省令は、品目ごと(製品群ごと)に適用になるものであって、事業所総体に対してのものではないという点も挙げられるでしょう。 QMSの調査は、QMS適合性調査申請に基づき実施されますが、これは対象製品(群)ごとに実施されます。 ご相談
よくある失敗と、外部の助言の活用の有用性
インターネット上に載っている都道府県等の雛形のまま製造販売業許可申請をすると、都道府県では問題なしとして認めることがあります。 しかし、実際にこれを運用するのは皆様の会社です。 許可取得後、QMSの調査を受ける際(認証機関等が実施)、会社の実情との乖離が大きい、運用がされていない、QMS省令の理解をしていないなどの理由により、数多くの指摘を受けるケースが散見されます。これにより医療機器の認証を受けるまでのスケジュールを大きく見直さなければならなくなることもあります。 弊所にも、そのような時点で初めてご相談いただく事例があります。 QMSの体制構築と製造販売業許可申請、製品の認証申請等は、一連の計画を立て、計画に則りタスクを実行してゆかねばなりません。 また、自社だけで取り組むこと自体は経験の蓄積になる面はあるのですが、第三者の目が入らないために許認可取得後のリスクについて見落としているケースもあります。 許可や認証の取得は目的ではなく、スタート地点に立つにすぎません。 許可業者として医療機器の認証等を維持して適切に法令を遵守して製造販売をするためには、計画段階から外部の目を入れ助言・支援を受けることをお勧めします。 よい行政書士や支援者であれば、その支援の結果が会社の無形の財産となるでしょう。 (品質マニュアルは雛形のままなどであれば会社の財産にはなりづらいです)
QMS・GVPは企業の法的リスクマネジメントでもあります
経営上の課題 製造物責任予防+事後対策
医療機器は、利用者、患者、医療関係者等の健康被害に直結する可能性があるものです。 ですから、医療機器の製造販売にあたっては、その事業者に製造管理・品質管理、安全管理ができる体制整備と、基準にしたがった運用管理が求められています。 事業者は、各種手順書等を作成したり、社内教育をしたりして、一定レベル以上の業務ができるよう図るのです。 医薬部外品で、白斑や、加水分解コムギ末による健康問題が生じ大きなニュースになりました。あれらの問題は、GVP省令とも密接に関連しています。 (医薬部外品の製造販売業者にも、製造販売後安全管理の基準としてGVP省令が適用になっています) 医療機器に関する法律には、薬機法のほかにもさまざまありますが、たとえば製造物責任法や民法(不法行為や債務不履行等)が関係します。 法令に従った適切な手順書に基づく運用は、業務の質を向上させるとともに、企業の法的リスクをあらかじめ低減してくれるでしょう。 万が一、医療機器に起因すると思われるトラブル(法的な問題)が生じた場合には、会社が法規制を遵守し対応してきたことを示すすべになるかもしれません。 QMS省令もGVP省令も、現場の担当者の方の業務だけでなく、経営層の方の責任について明示しています。省令の要求事項は、ただ許可証を得るために必要なハードルなのではなく、経営層の責務も含めた会社の業務の基準であるとお考えいただくとよいでしょう。 QMS・GVPの運用は、「製造物責任予防」だけでなく、「製造物責任防御対策」(証拠資料の生成・保管)になるものでもあるとお考えいただければと思います。 ※こうした観点からも、モデル手順書そのままであることはかえってリスキーだといえるでしょう。
法的リスクの3つの側面+α
損害賠償責任等(民事)
薬機法違反による刑事罰(刑事)
回収命令 業務停止、虚偽申請による許可取消等(行政)
医療機器の製造販売業者や製造業者は、これらの法的リスクを低減する方策を実施することが必要です。
適切な手順書を整備・改訂し、実施してゆくことが会社を守ることになるといえるでしょう。
※ 行政書士は、こうした法的リスクも勘案しながら、各種手順書を作成します。
ご相談

