医療機器の製造販売・製造の許認可申請
医療機器を製造販売・輸入するために・・・
2014年11月25日に医薬品医療機器等法(「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律」)が施行されました。この法律は従来の「薬事法」が内容と名称を変更されたものです。これにより、医療機器に関する許認可も大きく変わりました。
医療機器とは?(定義)
医療機器とは、次のようなものをいいます。
医薬品医療機器等法(旧称薬事法)第2条第4項
人若しくは動物の疾病の診断、治療若しくは予防に使用されること、又は 人若しくは動物の身体の構造若しくは機能に影響を及ぼすことが目的とされている機械器具等で、政令で定めるもの医療機関向けのものや治療目的のものに限らないことに注意して下さい。
- メス、注射器、医療用ガーゼなどから、医用X線診断装置などの医用電気機器、分析機器まで、医療機器には、さまざまな形状、用途のものがあります。
-
診断目的等で用いられるソフトウェアも医療機器に
→ 旧薬事法では、無体物であるソフトウェアは(日本では)医療機器には当たりませんでしたが、新法(医薬品医療機器等法)では、疾病の診断、治療、予防等に用いられるソフトウェアの場合、医療機器としての規制を受けることになりました。医療機器に該当するものであるか否かについては、個別に通知等に従い検討する必要があります。
-
「美容機器・健康器具だから医療機器ではない」?
→ 医療機器の定義に該当するものならば、「医療機器」にあたります。 美容機器と名乗りさえすれば医療機器に該当しないというわけではありません。ご注意下さい。
(美容機器、健康器具というのは医薬品医療機器等法上の区分ではありません)
美容機器(美顔器等)で認められる広告表現は、(事実である限り)化粧品で認められる効能効果の範囲内です。
医療機器の製造販売には、製造管理・品質管理・安全管理の責任が求められる
医療機器を自社製品として市場に流通させることを、上市といったり、製造販売といったりします。
製造販売する立場の会社は「医療機器製造販売業者」といいます。製造販売元、元売といわれることもあります。
患者、使用者の安全を守り、また、医療上の有効性を担保するため、医薬品医療機器等法は、医療機器を製造販売する会社に対し、医療機器の品質を保証し、安全を管理する責任を求めています。
医療機器の製造販売のためには、事業者が「医療機器製造販売業許可」の取得が必須です。
QMS省令という基準に従って製造管理・品質管理を行い、GVPという基準に基づき安全管理ができる体制が整っていることが、許可取得の要件になっています。
製造販売業者は、自ら製造所を持っていることもありますが、必ずしも自社製造である必要はありません。
製造所・工場が子会社(別法人)であってもよいですし、専門の技術を持つ製造業者に製造委託することも可能です。
(以下、「製造販売」と記してある部分は輸入も含みます)
製造販売業の説明部分(当ページ内4項)にジャンプします。
医療機器の製造行為には製造業登録が必要
医療機器を製造する事業者のうち、「設計」(*)、「主たる組立」「滅菌」「最終製品の保管」を行う事業者は、「医療機器製造業登録」が必要です。
事業所が国内である場合は当然ですが、外国からの輸入や、外国で設計しているような場合には、当該外国の事業所について、「外国製造業者登録」が必要です。
登録の取得は、要件を満たす責任者「責任技術者」を実地管理できるよう配置すること等が要件となっています。構造設備の適切さは、登録要件にはなっていません。(**)
したがって、旧薬事法時代から見れば、医療機器製造業登録は取得しやすくなったと言えるでしょう。
しかし、医療機器の製造にあたって、その医療機器の製造管理のために必要な設備、器具、作業環境等が求められることに変化はありません。設備器具、作業環境等については、QMS省令という製造管理・品質管理の基準の中に規定があり、原則としてこれに従って管理されることになります。
(適用が除外されている製品を除きます)
詳細は6項をごらんください)
製造業の説明(当ページ内6項)にジャンプします。
(*)クラス1医療機器のみの設計を行うのであれば、製造業登録は不要です。また、製造販売業許可の事業所で設計を行う場合も、登録不要です。
(**) 従来の薬事法では、医療機器製造業許可(外国医療機器製造業者認定)という制度でしたが、構造設備の要件が緩和されました。
医療機器を販売、譲渡、業として貸与する場合にも許可が必要です
医療機器製造販売業許可は、製造販売業者として医療機器を市場に流通させるために必要な許可ですが、人体へのリスクが一定以上ある医療機器を医療機関や消費者等に販売、譲渡、貸与(レンタル)等することについても、医療機器製造販売業許可とは別に、販売のための許可や届出が必要です。
医療機器の製造販売元がどこであるかにかかわらず、リスクの高い「高度管理医療機器」に指定されている医療機器や、保守点検に専門的な知識や技能を必要とする「特定保守管理医療機器」を、一般ユーザーに販売したり譲渡、賃貸したりする場合には、「高度管理医療機器等販売業(貸与業)許可」が必要です。
また、リスクが比較的低い「管理医療機器」を一般ユーザーに販売するには、「管理医療機器販売業(貸与業)届」が必要です。
医療機器を自社名義で製造販売するためには、複数の業許可・認定が必要なほか、
医療機器自体が、医療機器に要求されている基本要件、品目固有の要求事項(JISやIECなどが引用されている例が多いです)への適合が必要です。
高度管理医療機器等販売業・賃貸業許可についての説明ページへのリンクです。
医療機器の”クラス”を確認しましょう
医療機器のリスクに応じた「クラス分類」とは
医療機器(医療用具)には、人体(動物も含む)に直接触れるもの、体内に留置するもの、検査・分析用のものなどさまざまな用途のものがあり、人体等に対する危険度が異なります。
医療機器は、危険度により、クラス1(低)~クラス4(高)までの4種類に分類されています。
| クラス分類 | 薬機法上の 分類 |
リスク | 例 | |
|---|---|---|---|---|
| クラスIV | 高度管理 医療機器 |
きわめて高 | 患者への侵襲度が高く、 不具合が生じた場合、 生命の危機に直結する恐れが あるもの |
・植込み型 心臓ペースメーカー ・人工心臓弁 |
| クラスIII | 高 | 不具合が生じた場合、 人体へのリスクが比較的高い と考えられるもの |
・人工呼吸器 ・視力補正用 コンタクトレンズ |
|
| クラスII | 管理 医療機器 |
中 | 不具合が生じた場合でも、 人体へのリスクが比較的低い と考えられるもの |
・汎用針付注射筒 ・家庭用電気 マッサージ器 |
| クラスI | 一般 医療機器 |
低 | 不具合が生じた場合でも、 人体へのリスクが極めて低い と考えられるもの |
・ピンセット (単回使用でないもの) ・患者台 |
クラス分類表で確認
医療機器を製造販売したり、販売・賃貸等をしたりするにあたっては、クラスによって、必要となる許可や手続きが異なります。
したがって、まず、取扱予定の医療機器のクラス分類を確認する必要があります。
また、危険度は低くても保守管理に専門知識が必要な医療機器は、「特定保守管理医療機器」に指定されています。既存の医療機器(医療用具)は、国が示した「クラス分類表」に掲載されています。
扱おうとする医療機器の構造、使用目的、効能効果、などと、クラス分類表の類別、定義とを照合して下さい。
一般的名称ごとに、認証基準・承認基準・承認審査ガイドラインが定められていることもありますので、医薬品医療機器総合機構のページ(右記)でご確認下さい。
薬機法許認可の仕組みの制度・諸規制を理解しましょう
自社で医療機器を製造(製造委託する場合も含む)したり、輸入したりして、自社の名前で市場に流通させる(製造販売)には、医薬品医療機器等法に基づく「医療機器製造販売業許可」が必要です。
医療機器製造販売業許可は、取扱医療機器のクラスに応じて第1~3種に分かれています。
また、クラス3、4に該当する医療機器や、特定保守管理医療機器を、医家やユーザ等に販売・譲渡・賃貸するには、医療機器製造販売業許可とは別に、「高度管理医療機器等販売業・賃貸業許可」が必要です。
(高度管理医療機器販売業・賃貸業許可を持っている販売業者、製造販売業者、製造業者だけに譲渡する場合は不要)
クラス2の医療機器の販売(特定保守管理医療機器以外)の販売には、「管理医療機器販売業届」が必要です。
医療機器を製造するためには、製造所ごとに製造業許可が必要です。
医療機器を製造販売(輸入を含む)する場合の規制
医療機器のクラスに応じて必要な許可が異なります。
| 規制対象 | 製造販売業者 | 医療機器(製品) | 販売店・営業所 |
|---|---|---|---|
| 医療機器 | |||
| クラスIII、IV | 第1種 医療機器製造販売業 許可 |
承認 | 高度管理医療機器等 販売業許可 |
| クラスII (認証基準(*)なし/ 基準に適合しない) |
第2種 医療機器製造販売業 許可 |
承認 | 管理医療機器 販売業届 (特定保守管理医療機 器を除く) |
| クラスII (認証基準あり &基準に適合する) 「指定管理医療機器」 |
第2種 医療機器製造販売業 許可 |
認証 | 管理医療機器 販売業届 (特定保守管理医療機 器を除く) |
| クラスI | 第3種 医療機器製造販売業 許可 |
届出 (新医療機器は 承認取得が必要) |
– |
第1種医療機器製造販売業許可を取得すると、クラス3、4だけでなく下位のクラス(1,2)の医療機器も製造販売可能です。
第2種医療機器製造販売業許可を取得すると、クラス1の医療機器も製造販売可能です。
医療機器の製造所、保管場所等の規制
医療機器を製造する事業所は、担当する工程により、医療機器製造業登録、または外国製造業者登録が必要な場合があります。
旧法に比べ、許可制から登録制になり、また構造設備要件が緩和される等、新規参入はしやすくなりました。
他方、製造業者は、QMS省令に基づき製造所の品質マネジメントシステム(QMS)を構築する必要があり、QMSの体制構築は製造販売業者による第二者監査や審査機関による審査の対象となります。
構造設備は都道府県の審査する許可要件ではなくなったものの、引き続きQMS省令の関連する規定に基づき適切に運用管理されなければなりません。
| 製造行為 | 設計 | 主たる組立て | 減菌 | 国内 最終保管 |
|---|---|---|---|---|
| 製造対象 | ||||
| プログラム | 〇 | - | - | - |
| プログラムの 記録媒体 |
〇 | - | - | 〇 |
| クラスI | - | 〇 | 〇 | 〇 |
| 上記以外 (プログラム以外の クラスII、III、IV) |
〇 | 〇 | 〇 | 〇 |
医療機器製造販売業許可とは
医療機器の製造販売にもとめられる品質保証・安全管理体制
医療機器を市場流通させる事業者(製造販売業者。輸入業者もここに含まれます)は、医療機器の製造販売業許可を取得しなければなりません。
医療機器を日本国内市場に上市するにあたっては、医療機器の品質を保証し、ユーザーや患者、医療関係者等の安全を管理する責任を負います。
そのため、薬事法では、製造販売業許可の要件として、製造管理・品質管理と安全管理の体制を整えることを求めています。
医療機器製造販売業許可は一法人にひとつの許可です。
(第1種医療機器製造販売業許可と第3種医療機器製造販売業許可を同時に持つことはありません)
総括製造販売責任者(後述)の勤務する事業所(本社等)がある都道府県知事に、許可を申請します。
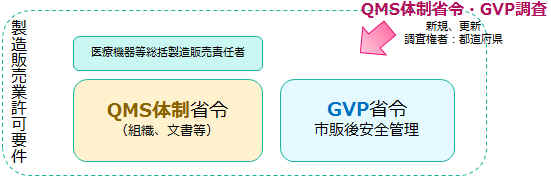
医療機器等総括製造販売責任者[2012年8月30日 要件改正]
医療機器の製造販売を総括する責任者です。
下記の資格要件を満たす方を常時配置してください。
常勤で配置する。
国内品質業務運営責任者、管理監督者、管理責任者と(支障がない限り)兼務可能。
[総括製造販売責任者の資格要件]
(医薬品医療機器等法施行規則114条の49)
-
第1種、第2種 下記のいずれか
1.大学等で物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学、歯学のいずれかの専門課程を修了
2.旧制中学若しくは高校又は同等以上の学校で物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学、歯学のいずれかの専門課程を修了後、医薬品、医療機器又は再生医療等製品の品質管理又は製造販売後安全管理に3年以上従事
3.医薬品、医療機器又は再生医療等製品の品質管理又は製造販売後安全管理に5年以上従事後、総括製造販売責任者講習会を修了
4.上記と同等の知識経験があると厚生労働大臣の認めた者
-
第3種 下記のいずれか
1.旧制中学、高校又は同等以上の学校で物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学、歯学のいずれかの専門課程を修了
2.旧制中学、高校又は同等以上の学校で物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学、歯学のいずれかの科目を修得後、医薬品、医薬部外品、化粧品、医療機器又は再生医療等製品の品質管理又は製造販売後安全管理3年以上従事
3.上記と同等の知識経験があると厚生労働大臣の認めた者
国内品質業務運営責任者
品質保証部門の責任者です。
[国内品質業務運営責任者の基準]
品質保証部門の責任者品責には医療機器、医薬品又は再生医療等製品等の許可業者における3年の品質管理等の実務経験が求められる点に留意してください。(限定第3種における経験は限定第3種の申請等にのみ認められます)
[製品リスクを勘案し、業務経験を十分有する等、関係業務を熟知した者であるべき]とされています。
「その他これに類する業務」とは、
総括製造販売責任者
製造管理者
輸入管理者
品質管理責任者
製造管理責任者
等とされています。
第2種、第3種の場合は、次のような方も含まれます。
ISO9001又は13485の認証を取得している製造業者(医療機器に限らない)における品質マ ネジメントシステムの維持管理の経験、又は内部監査の経験 3年以上
安全管理責任者
安全確保業務の責任者です。
[安全管理責任者の基準]
[第1種のみ]安全管理統括部門の責任者 [第1種のみ]第1種の場合は、安全管理統括部門を置き、その責任者であること、
また、安全確保業務に3年以上従事した経験が求められます。
第2種、第3種は、3年の経験は求められていません。
「その他これに類する業務」は、市販後調査責任者の業務等があたるとされていますが、医薬情報担当者、医療機器情報担当者としての業務のみでは認められないとされています。
製造管理・品質管理体制、安全管理体制の構築(6参照)
国は、製造管理・品質管理の体制に関する基準(QMS体制省令)、医療機器の市販後安全管理に関する基準(GVP省令)を、省令として公布しています。
医療機器の製造販売業許可業者は、これらの省令に遵って、適切に、医療機器の製造管理・品質管理の体制を整備し、また、QMS省令・GVP省令に基づき業務を行わねばなりません。
QMS体制・GVP体制が整っていることが、「許可の取得」「許可の維持」の要件です。
医療機器の製造販売において、最も基本的な用語ですので、ぜひ覚えてください。
製造管理・品質管理の基準(QMS)、安全管理の基準(GVP)の解説ページです
QMS/GVPの体制構築(組織、手順書、記録類)
医療機器製造販売業者は、QMS、GVPの体制構築が必須要件
医療機器製造販売許可業者は、事業の遂行のために、手順書を整備し、製造・包装・出荷・試験検査・情報収集・報告などの記録をつけるなど、手順書による運用をすることが必要です。
医療機器製造販売許可業者は、QMS体制省令・QMS省令に基づき、「品質管理監督システム基準書」や下位の各種文書、記録類を作成する義務があります。
また、GVP省令に基づき、市販後安全管理体制を整えておかねばなりません。
第3種医療機器製造販売業者は、安全管理”手順書”の作成義務はありませんが、適正な業務遂行のためには、手順書の整備は事実上必須といえます。
QMS、GVPの体制の整備は医療機器製造販売業の「許可要件」に、QMSへの適合は医療機器の製造販売承認や認証の取得の要件になっていますから、
医療機器の業許可申請や医療機器の承認・認証申請のときまでには、手順書や記録類、責任者や担当者の決定、「具体的に作りこんだ手順書」、運用できる体制づくりが必要です。
製造管理・品質管理の基準(QMS)、安全管理の基準(GVP)の解説ページです
QMS・GVP手順書は、実態に合ったものを・・・
都道府県では、審査時に用意されている手順書が適切であるかどうか、その内容を事業者が理解して、適切に運用できる状況かどうかなどを、実地調査で判断します。
申請段階ではもちろんまだ事業はスタートしていませんが、取り扱い医療機器、会社の組織、販路等に応じ、許可取得後適切に運用できるような手順書・組織になっていることが、新規申請時の鍵です。
当事務所のご相談・ご依頼案件の中には、自ら申請をされた事業者のかたで、QMS・GVP手順書が不適当なためになかなか県庁のOKをもらえず、許可取得がなかなかできないも多く見受けられます。
なかなかOKがでませんと、当初予定した時期に事業がスタートできないようなことにもなりかねません。
ですから、初期段階で、教育訓練を通じて、薬機法許認可を知り、QMS・GVPの要求事項をよく理解して、
サプライヤー・製造所との関係、扱う医療機器の特性、社内組織等を踏まえ、実のある手順書を整備することが事実上必須だといえるでしょう。
当事務所では、新規申請や許可更新等に際し、教育訓練の一環として、役員/ご担当者様に薬事法・QMS・GVPをご理解いただくことに主眼を置いた教育訓練(社内勉強会)の機会を設けております。
当事務所でQMS・GVP手順書・記録様式整備の支援を行っております。
許可の更新
医療機器の承認・認証を維持し、また医療機器製造販売業許可や製造業登録を更新するためには、QMS、GVP体制の整備やQMS省令への適合、品目ごとの記載整備などの諸手続きが必要です。
初めての許可取得時と異なり、前回の更新以降にQMS・GVPが適切に運用されてきたかどうかを調査されます。
審査段階でQMS・GVPの運用が十分でない、という状況になりますと、場合によっては許可期限を過ぎても更新できなかったり、製品の承認・認証の効力を欠いたりすることもあります。
下記のページをご参照の上、事業に支障の出ないよう準備されることをお勧めします。
許可の更新について 詳細はこちらをご覧下さい。
なお、当事務所では、「内部監査・自己点検」の支援をしております。簡易的な監査を当事務所で行う、あるいは、貴社が行う自己点検等を支援する業務です。
これを適宜行うことで、問題点の是正や、よりよい業務方法に改善することができ、円滑な更新だけでなく、業務の質の向上、薬事法対応力の許可に繋がります。
医療機器製造販売業・製造業等の許可更新のポイントなどを解説しています。
医療機器製造業登録とQMS
医療機器製造業登録の要件
「責任技術者」の配置が重要な要素です。
旧法の時代には「構造設備規則」への適合を審査されましたが、現行法では、構造設備はQMS省令の構造設備関連規定への適合を製造販売業者やQMS調査権者により確認されます。
[責任技術者の資格]
下記のいずれか
-
原則 下記のいずれか
1.大学等で物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学、歯学のいずれかの専門課程を修了
2.旧制中学若しくは高校又は同等以上の学校で物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学、歯学のいずれかの専門課程を修了後、医療機器の製造に3年以上従事
3.医療機器の製造に5年以上従事後、責任技術者講習会を修了 上記と同等の知識経験を有すると厚生労働大臣が認めた者
-
クラス1のみ製造する場合(緩和)
1.旧制中学若しくは高校又は同等以上の学校で物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学、歯学のいずれかの専門課程を修了
2.高校で物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学、歯学のいずれかの科目を修得後、医療機器の製造3年以上従事
3.上記と同等の知識経験を有すると厚生労働大臣が認めた者
-
設計のみ行う場合
前2項に関わらず、製造業者が設計部門の責任者として指定する者
製造所におけるQMS省令の準用
QMS省令第6章(83-84条)により、医療機器製造業者においても適切なQMSの構築が必要です。
製造する製品についてその製造業者で行う工程に照らし、適用することが適当ではないQMS省令の要求事項については、製造業者の品質管理監督視システムに適用しないことができます(QMS省令85条)。
また、製造販売元は、製造業者が適切なQMSを構築していることを確認しなければなりません。
医療機器の製造においては、製造業登録を受けるのみならず、QMSの構築も必要となるという点にご留意ください。
当事務所では、製造所のレイアウトや識別表示などについて、QMS適合性も踏まえて適否・助言等を行っています。
医療機器製造業者,医療機器の承認・認証に必須のQMS省令について、簡単にご説明しています。
医療機器の設計開発と承認、認証、届出
医療機器ごとに承認・認証・届出が必要
医療機器の販売のためには、業許可だけでなく、医療機器ごとに承認、認証を取得したり、届出を行ったりしなければなりません。
承認申請書・認証申請書・製造販売届書は、設計開発関連の事実が反映されるものです。
| クラス4 | 国による製造販売承認 |
| クラス2,3 ・認証基準に適合しないもの ・認証基準が存在しないもの |
国による製造販売承認 |
| クラス2,3 ・認証基準に適合するもの |
国から指定をうけた第三者機関による認証 |
| クラス1 | PMDAへの製造販売届出 |
| クラス1のうち、 新規性のある医療機器 |
国による製造販売承認 の後、(承認整理・)届出 |
届出は提出のみで手続きが完了しますが、その他は独立行政法人医薬品医療機器総合機構(PMDA)又は認証機関による審査があります。
詳細は、別途ご相談ください。
基本要件への適合
医療機器は、最も基本的な要求事項である基本要件(平成17年3月29日厚生労働省告示第122号)に適合するように設計・開発されていなければなりません。
この基本要件では、設計開発上の要求事項(電気的、化学的、物理学的、生物学的安全性など)に適切に適合する形での設計開発が要求されています。
医療機器は、後述するように、品目毎に認証や承認、届出が必要ですが、いずれの医療機器も基本要件に適合していなければなりません。
特に、クラス2以上の医療機器の場合、認証や承認の審査段階で、基本要件に適合している証拠資料の提出を求められます。
また、設計段階で、JIS T14971(ISO 14971)に基づくリスクマネジメントも実施しておくことが必要です。(これが設計をする上でのインプットにもなります)
医療機器の設計開発に当たっては、基本要件やJIS規格などをよく確認し、承認申請・認証申請で求められる記載事項や証拠資料なども念頭に置いて下さい。
指定管理医療機器、指定高度管理医療機器の認証
管理医療機器(クラス2)及び高度管理医療機器(クラス3)については、認証基準が国から告示されています。
認証基準に適合するものを、指定管理医療機器、指定高度管理医療機器といいます。
指定管理医療機器・指定高度管理医療機器の製造販売にあたっては、国が指定する民間の認証機関に対して「認証」を申請し認証を取得する必要があります。
認証基準において、基本要件チェックリストが公表されており、また、適用となるJISも明確化されていますので、設計開発にあたってご参照ください。
認証基準から逸脱するものは、厚生労働大臣から承認を取得することになります。
輸入品の場合の留意点
海外で流通している医療機器を輸入使用とする場合、その医療機器が日本の基本要件やJIS等に基づいて設計開発されているわけではないことが多いでしょう。
そのため、基本要件や認証基準への適合、先発品との比較検討、物性・生物学的安全性、その他の試験を改めて実施しなければならないことも多くあります。
また、承認品目で、後発医療機器(同一/実質的同等)、改良医療機器、新医療機器にあたるかの判断にあたって必要な情報を、日本側が入手しておくことも必要です。
外国製造業者登録/輸入届
国内で流通させる医療機器の製造所で、設計・主たる組立・滅菌を行う外国の製造所は「外国製造業者登録」を受ける必要があります。
また、扱い医療機器がクラス2以上の場合には、医療機器の承認・認証申請の際に、当該外国製造業者に対してQMS適合性調査が行われますので、QMS省令の基準にも合致している必要があります。
医療機器製造販売業許可を取得した後は、輸入をするために、7で述べた承認、認証、届出のほか、「製造販売用医療機器輸入届書」の提出が必要です。
会社の設立
医療機器の製造販売業は、会社を設立して行われることをお薦めします。
まず会社を設立し、その会社が許可申請し許可を取得するという流れになります。
個人事業で医療機器製造販売業許可を取得した場合は、製品には、「屋号」のほかに「個人の姓名」を表示することになります。
個人で許可を得て事業をされていらっしゃる事業者様もありますので、これを全く否定するものではありませんが、多くの場合は、
といった観点から、会社を設立されることが多いようです。
個人事業の場合、許認可は事業主に帰属していますので、事業主死亡の場合に、事業継続に支障をきたすことになります。
もちろん、個人事業でも会社でも、同じように品質保証・安全管理の体制を整えて事業を行うことに違いはありません。
会社設立の詳細は、こちらをご覧ください。
会社設立の基礎知識の解説ページです。

